软件教程
来自我们的高级技术支持专家,精心撰写整理的原创学习教程
原创精选
便捷搜索
快速入门和提高
教程(二十五)使用赝势基组时5d-6d基函数一致性问题
发布时间:2021-06-08
关键词:赝势 基组 5d-6d 基函数 一致性
(一)引言:
在含有金属的体系中,如金属有机化合物、金属氧化物的计算中,由于相对论效应和计算量的问题,我们通常用赝势基组对金属元素进行描述,而对其它非金属元素则使用全电子基组(例如6-31G(d))。这个看起来很完美的混合基组策略,由于Gaussian程序的默认设置而隐藏了一个很严重的科学问题。这个问题涉及到基函数的组成问题,Gaussian程序中一般默认使用6d关键字(但在使用gen或genecp等关键字时往往默认为5d)。6d代表使用6个高斯函数拟合d轨道,5d代表使用5个高斯函数拟合d轨道。正是因为这个原因,会让很多做类似体系的研究者“自然地”得到了错误的结果。这个错误结果与分子大小、结构有关,有时偏离正确值甚至会达到100 kcal/mol。本文将以Fe(H2O)3+6与CO2相互作用为例介绍在使用混合基组时,如何正确地设置基函数,得到正确的结果。
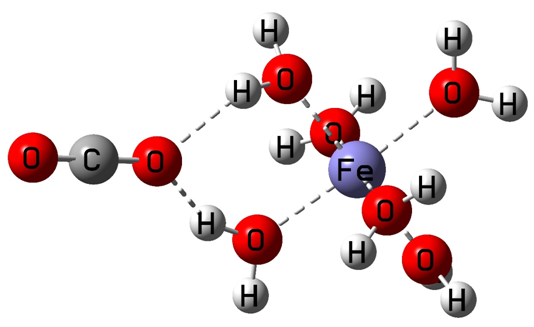
(二)计算过程:
1. 单体优化及频率计算
软件版本Gaussian 16, GaussView 6;
模型化学:B3LYP/6-31G(d), B3LYP/genecp,empiricaldispersion=GD3,使用6d基函数;
首先进行CO2单体的优化,由于分子较为简单,此处就省略建模过程。对于CO2分子,使用B3LYP/6-31G(d)方法,此方法默认为6d基函数,所以无需特别指定。输入文件如下:
注:empiricaldispersion关键词无需全部写出;
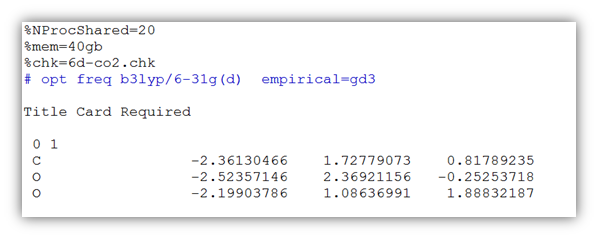
其次,进行Fe(H2O)3+6单体的优化,如引言所述,此处的设置较为关键。此时,如果使用genecp关键词,则需要指定使用6d基函数,输入文件内容如下:
%NProcShared=20
%mem=40gb
%chk=correct-5d-Fe-6h2o.chk
#opt freq b3lyp/genecp empiricaldispersion=gd3 6d
TitleCard Required
3 6
Fe -0.81296853 -0.18041906 -0.19261956
O 0.78569716 0.66806818 0.07037726
H 1.48104572 0.03883428 0.23517534
O -1.55939731 1.36575711 -0.77382096
H -0.97746026 2.10053407 -0.57446137
O -0.23509270 -0.73651493 -1.83490138
H -0.30279566 -1.71362870 -1.89886383
O -2.38210141 -1.04528474 -0.48165950
H -2.63186070 -1.51423581 0.31941981
O -1.38052731 0.37221423 1.42457516
H -2.14955685 -0.13284946 1.67194119
O -0.28879156 -1.72328564 0.62601854
H -1.03589238 -2.12812110 1.07800682
H 0.71494426 1.26121184 0.84758713
H -2.40351697 1.50449797 -0.38792184
H 0.69269100 -0.50062637 -1.95375612
H -1.59976933 1.30253499 1.38951928
H 0.06269552 -2.33629946 -0.02700722
H -2.27591361 -1.65713170 -1.20154960
H O 0
6-31G(d)
****
Fe 0
Lanl2dz
****
Fe 0
Lanl2dz
Fe(H2O)3+6单体结构如下:
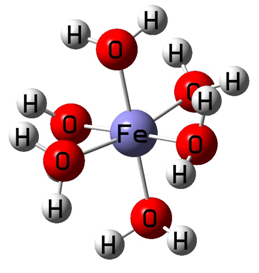
输入文件设置完成之后,投入Gaussian程序进行计算。
打开log文件,在赝势参数说明前有一句话:
General basis read from cards: (6D, 7F)
说明此时已经使用6d基函数拟合d轨道。
2. 复合物结构优化及频率计算
构建Fe(H2O)3+6与CO2相互作用模型,设置基函数的类型为6d,输入文件如下:
%NProcShared=20
%mem=40gb
%chk=correct-5d-Fe-6h2o-co2.chk
#opt freq ub3lyp/genecp empiricaldispersion=gd3 6d
Title Card Required
3 6
Fe 0.40509700 -0.00055900 0.00005000
O -0.93618500 -0.64463000 1.39156500
H -0.73820800 -0.91515300 2.31533800
O 0.12814300 1.91987400 0.62126600
H -0.47117300 2.22284600 1.33874700
O 1.91397700 -0.22085300 1.35113300
H 2.45369100 -1.02848500 1.50007300
O 1.74625200 0.64247200 -1.39161700
H 1.54790400 0.91294700 -2.31533200
O -1.10394100 0.21938200 -1.35030100
H -1.43100500 -0.46117300 -1.97929800
O 0.68148600 -1.92075400 -0.62141300
H 0.24216900 -2.72235700 -0.26061500
H -1.90620900 -0.74292200 1.26850600
H 0.56810600 2.72150300 0.26131800
H 2.24053400 0.45909700 1.98104500
H -1.64409000 1.02676100 -1.49909500
H 1.28102900 -2.22388600 -1.33864600
H 2.71603800 0.74288200 -1.26824200
C -2.36130466 1.72779073 0.81789235
O -2.52357146 2.36921156 -0.25253718
O -2.19903786 1.08636991 1.88832187
C H O 0
6-31G(d)
****
Fe 0
Lanl2dz
****
Fe 0
Lanl2dz
复合物结构如下:
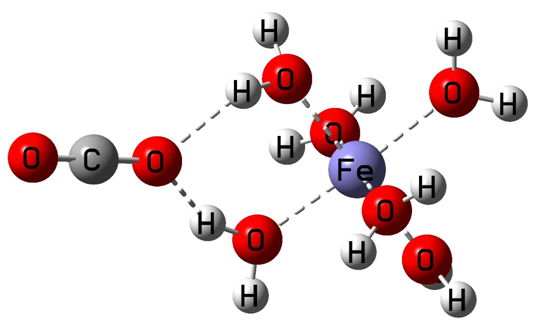
输入文件设置完成之后,投入Gaussian程序进行计算。
3. 相互作用能计算
待上述3个作业完成之后,摘录各个结构的焓值(H),得到相互作用能:
CO2 (a.u.) | Fe(H2O)3+6(a.u.) | Complex(a.u.) | ΔE (kcal/mol) |
-188.565949 | -580.755842 | -769.35585 | -21.4 |
按照上述计算方式才是正确的使用混合赝势基组的方法。下面列举一个常见的错误用法。
4. 常见错误用法
对于Fe(H2O)3+6及与CO2形成的复合物,都不明确指定6d基函数,而使用默认的5d基函数(即上述输入文件中不特别指明6d)。投入Gaussian程序进行计算;
相互作用能计算:
CO2 (a.u.) | Fe(H2O)3+6(a.u.) | Complex (a.u.) | ΔE (kcal/mol) |
-188.565949 | -580.755842 | -769.35585 | -21.4 |
-188.565949 | -580.744906 (5d) | -769.341743 (5d) | -19.4 |
如上表,得到的作用能为-19.4 kcal/mol,比正确的作用能高2.0 kcal/mol。可见,如果错误使用混合赝势基组,将会对结果造成很大影响,尤其对于弱相互作用体系来说是致命性的。
值得注意的是,这个错误结果与分子大小、结构有关,有时偏离正确值甚至会达到100 kcal/mol。